Introduction :
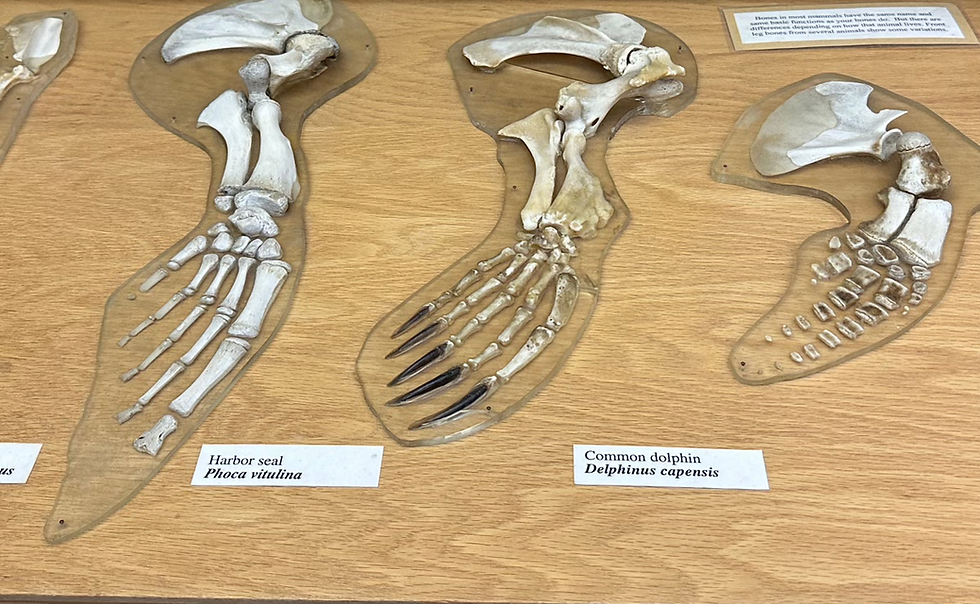
Fossils and Geologic records
Fossil and geologic records once served as the main temporal anchor point for understanding evolutionary timelines. (Donoghue & Benton, 2007). However, fossil evidence sources have limitations. These include gaps in the fossil record, taxonomic biases (the preservation of some taxa more than others), and stratigraphic uncertainties. Additionally, The age estimates of fossil occurrences may not represent the true origin of the group. The oldest fossil record of a lineage may only reflect the time when a stable population with diagnostic morphological traits became widespread enough to be preserved in the fossil record. Incorporating different phylogenetic approaches from multiple biological fields may enable a more comprehensive view of macroevolutionary patterns.

Comparative Morphology
Comparative morphology practices provide excellent insight into phenotypic relationships, revealing changes in form and function over time in extant taxonomies. However, researchers in this field face their own set of challenges. For example, convergent, or homoplastic evolution, is a potential source of error that can result in misleading phylogenetic inferences (Lee & Palci, 2015). This field also deals with human errors introduced via incomplete data sampling and the misidentification of species and characters, resulting in modeling inadequacies.
The molecular clock
Mid-century advancements in molecular biology shifted the focus towards molecular analysts to provide the biological world with answers to questions about the mechanistic basis that underlie organismal divergence. This presented new pieces of evidence to the puzzle of life on earth (Martinez, 2018.) In 1965, Zuckerkandl and Pauling introduced the idea of a molecular clock. Their research inferred a constant rate of amino acid substitutions over time. They compared this constant rate of change to the ''ticks'' of a clock (Zuckerkandl & Pauling, 1965). In 1968, Motoo Kimura introduced the ''neutral theory of molecular evolution''. He suggested that the clock reflects the action of random drift, not natural selection (Kimura, 1968). Today, the term 'molecular clock' describes genome sequence changes over time.
Nearly Neutral Theory
Ohta, In 1973, presented a modified version of neutral theory calling it the ‘nearly neutral theory’. She proposed a modified version of the neutral theory by stating that most genetic changes are neutral, but a small proportion is either beneficial or deleterious. The ‘nearly neutral theory’ allows for variable rates of evolution and changes in heterozygosity and has provided the framework for developing a relaxed-molecular clock model. Another major advance in molecular dating was 'The coalescent theory' a basis of statistical techniques that provide extensions of classical population genetics with mathematical models that reflect genomic data and biological influences (Kingman, 1982)

Total Evidence Dating
These modifications are widely used in population genetics because they account for variable substitution rates and population sizes. Based on the above mentioned research, Total Evidence Dating, a modeling methods that accounts for these uncertainties and acknowledges fossil evidence as an indicator of relative dating, Bridging fields of data, in ways that incorporate morphological and molecular, fossil and other relevant lines of evidence, reduces discrepancies creating more accurate divergence time estimateS
SIGNIFICANCE AND IMPORTANCE Of A '’BIG DATA’ APPROACH
The collaboration between fields of study and datasets has led to more informed conclusions and drawing more contextually meaningful inferences. However, despite advances in our ability to create accurate timelines of complex lineages, limitations, inaccuracies, and discrepancies remain prevalent. Recent studies have found the potential for substantial errors when substitution and speciation rates vary within lineages (Ritchie, Hua, & Bromham, 2022). Improvements in applied methodologies are needed in order to increase levels of fidelity measurements. This paper aims to identify both the challenges and innovations in phylogenetics that successfully merge unique datasets from different fields of science, each with its strengths and weaknesses, to form well-rounded conclusions.
We review recent research tools and approaches in molecular dating and phylogenetics by highlighting recent studies in phylogenetics that progress the combined effort of calibrating the molecular clock. The studies chosen utilize (or argue in support of, big data approaches, utilizing many lines of evidence (i.e., morphological, fossil record/stratigraphic, climate, and biogeographic) datasets and combine them with relative dating methods such as morphological and molecular. The following research studies (Hipsley & Muller, 2014; May et al., 2021; Fernández et al., 2017) demonstrate 'big-data' approaches to their phylogenetic analysis studies.

Collaborative efforts between scientific fields cultivate a deeper understanding of historic evolutionary relationships and influences through space and time. Understanding the past is a preface to being able to predict future outcomes. These are vital tools for conservation decision-making in a future filled with dynamic environmental change. Developing robust, accurate phylogenetic modeling tools is relevant to all fields of science conducting biological investigations.
STATE OF THE FIELD
A macroevolutionary lens and interdisciplinary knowledge are required to understand the events influencing earth's evolutionary history. By integrating molecular, phenotypical, morphological, and paleontological lines of evidence, estimations about biological systematics have improved significantly, providing a clearer picture of phylogenetic relationships. Advances in computational biology have increased our ability to develop increased accuracy when estimating divergence times. Key advancements include:
1) Phylogenomics: or genome-scale sequencing datasets are used in phylogenetic studies for analyzing relative rates of change in the genome over time. (Kumar et al., 2012) .
2) Coalescent theory models: a statistical approach to simulating the process of genetic drift and lineage coalescence over time and enabling the estimation of population parameters and the reconstruction of phylogenetic trees.(Kingman, 1982).
3) The multispecies coalescent model describes gene trees as independent random variables generated along the lineages of the species tree. Since the multi-species coalescent model allows gene trees to vary across genes, coalescent-based methods account for heterogeneous gene trees in phylogenomic data analysis (Jiao, Flouri, & Yang, 2021).
4) Bayesian Markov Chain Monte Carlo (MCMC) based algorithms account for flexible parameters, and applying prior distributions (anchoring calibration points provide temporal bounds for estimating divergence times among clades) combined with extant morphology data enables the exploration of the posterior distribution patterns. (Larget & Simon, 1999)
CASE STUDIES: CALIBRATION CHALLENGES IN RECENT RESEARCH

Beyond fossil calibrations: realities of molecular clock practices in evolutionary biology (Hipsley & Muller, 2014): This study, published in 2014, discusses the importance of accuracy and precision in divergence dating analyses and the roles involved in calibrating the molecular clock. Fossils are a way to date the molecular clock externally (absolute dating), while the molecular clock provides relative dating (Hipsley & MÃller, 2014). Variations in calibration methods resulted in the highest levels of discrepancies between divergence time estimates. The authors aimed to identify recent patterns in published clock calibration studies and analyze potential pitfalls associated with each methodology. To do this, they conducted a literature survey of 600 publications from 2007-2013. At the time of the study, they found that the most commonly used methods for absolute dating were led by fossil evidence (approximately 50%), geologic events, and secondary dating methods. The researchers in this study found that using fossil evidence as anchoring points is a potential source of error in estimation. In addition, due to the prevalence of taxonomic biases, many taxonomic groups were under-represented within the fossil record and therefore neglected in calibration implementation guidelines. The authors also warned against using geologic events alone to explain allopatric dispersal patterns suggesting that many clades may be older than assumed from geographic isolation evidence. Finally, they also discuss the strengths and weaknesses in molecular dating and the need for continued progress in developing standards for substitution and mutation rates per clade. 2007 marked the release of a revolutionary computational tool BEAST or Bayesian evolutionary analysis, by sampling trees. Its developers describe this tool as an "evolutionary analysis package for molecular sequence variation. It also provides a resource for further developing new models and statistical methods" (Drummond & Rambaut, 2007). In 2007 this novel tool enabled the incorporation of multiple sources of evidence and various statistical analysis models. The authors of this paper encourage the use of combined evidence methodologies for improved estimation, stating that "Age constraints based on other types of data provide alternative means that, when well justified, can contribute critical information on the evolutionary history of life."

Inferring the Total-Evidence Timescale of Marattialean Fern Evolution in the Face of Model Sensitivity' (May et al., 2021): This study found accuracy in the divergence time estimations of Marattialean fern evolution and attributed these results to two revolutionary developments in phylogenetics:
1) Total-evidence dating (TED)and 2) Fossilized birth-death (FBD) tree models. The developments of FBD models provide the statistical framework for implementing both paleontological evidence (stratigraphic data) with neontological (ie: molecular sequence data (Matschiner, 2019). In addition, total-evidence dating (TED) allows for including fossils as tips in the analysis rather than in the nodes-based dating. In TED, the tips represent the descendants of a taxa species, while the nodes represent the most common recent ancestors of the descendants. Before TED dating, phylogenies were dated by calibrating the interior nodes against fossil records. Unfortunately, this approach led to the misrepresentation of fossil records (Rodfnquist et al., 2012). This paper applies both TED and FBD models to the study of Marattialean Fern clades to develop a deeper understanding of their evolutionary history and the phylogenetic divergence times. The authors compared the results of different approaches. They placed fossil evidence at the tips of the tree and compared this data to results of implementation at the nodes of the tree. They found dramatic differences. The two approaches greatly influenced both divergence times estimates and topological relationships. The authors report that tip dating provided more accuracy and superior results. Their best-fitting models inferred that the stem group divergences, those of the extinct taxa of Marattiales", occurred in the mid-Devonian. The crown divergences (the most recent common ancestor of extant taxa) occurred in the late Cretaceous. The models indicated an elevated speciation rate in the Mississippian and an elevated extinction rate during the Cisuralian. Marattiales experienced its highest levels of diversity (approximately 2800 species) at the end of the Carboniferous, followed directly by a rapid decline which ultimately resulted in the extinction of the Psaroniaceae. Psaroniaceae descendants, the Marattiaceae, remained in a relatively stable state until the present day. The study warns about the challenges and inaccuracies that may result from utilizing node-based approaches (as opposed to tip-based) to estimate divergence time. Node-based approaches are influenced by the quality and completeness of the fossil record and can potentially lead to biased or inaccurate estimates of divergence times. On the other hand, TED or tip-dating approaches incorporate fossils directly into the analysis and can provide more accurate and reliable estimates of these parameters, mainly when the fossil record is relatively complete. The authors highlight the importance of choosing models that track morphological traits over time or explicit models of morphological evolution. These models consider a broader range of data that may affect how quickly or slowly species evolve and diverge from each other. By comparing the results of comprehensive models, researchers can accurately estimate the timing of significant events in the evolutionary history of a species.

The Opiliones tree of life shedding light on harvestmen relationships through transcriptomics (Fernández et al., 2017). The authors in this study explore relationships between clades of Opiliones, arachnids commonly known as huntsman or "daddy long legs." This study is the first to use a TED approach on Opiliones. The authors found the model to be superior due to its ability to remove the guesswork from fossil placement within the tree. The study combines fossil evidence, biogeographical dating, molecular dating, and transcriptomics to calibrate their phylogenies. Transcriptomics is the study of RNA sequencing and transcriptomes. Transcriptomes are complete sets of cell transcripts. Transcriptomic researchers work to identify/quantify transcriptomes because they indicate specific developmental stages and physiological conditions in the sample (Wang, Gerstein, & Snyder, 2009). This methodologies in this research employ a similar approach to the Marattialean fern study. Both papers utilize a TED approach and report the benefits of incorporating fossil evidence in tips versus nodes. According to the authors, fossils can either constrain specific nodes in the phylogeny (node dating) or serve as terminals in a combined analysis of morphology and molecules (total evidence dating). The accuracy of these methods depends on the placement of fossil material. One of the benefits of the TED approach is its ability to incorporate any uncertainty in this line of evidence into calibrations. It treats all well-characterized fossil species as terminals and allows for explicit parameterization of the point of divergence for a given fossil (along a branch using a morphological data set). The study emphasizes the importance of taxonomic sampling in molecular dating, particularly for diverse clades like arthropods.
OUTSTANDING QUESTIONS & OBSTACLES TO PROGRESS :
The tools used interdisciplinary approaches, mathematical modeling, and computational biology have significantly improved our methods for calibrating the molecular clock. Despite these advancements, there are still inherent limitations in our models. It is important to remember these mechanistic models are just the tools we use to explain biological systems. They are also limited by the quality of the data implemented, and the appropriate choice of statistical models applied. One bottleneck worth mentioning is not just the quality of the data we use, but the openness and transparency of the data's accessibility. Increased transparency and replicability in our research data will lead to improvements in the value of our scientific inferences. Unfortunately, inaccuracies and inadequacies in research reporting methods have hindered the field's advancement and prevented the use of the data in meta-analysis studies. There are several strategies for increased transparency such as mandating data sharing, pre-registration of studies, and conducting replications can improve transparency and data quality (O'Dea et al., 2021).
Access to genome sequencing data and the use of modern statistical techniques has improved the molecular clock calibrations. However, the field can further mature by fostering collaboration between different disciplines, and testing new combinations of methods. These practices will develop a higher-resolution view of evolutionary history.
CITATIONS:
Brown, J.W. and Smith, S.A. (2018) ‘The Past Sure is Tense: On Interpreting Phylogenetic Divergence Time Estimates’, Systematic Biology, 67(2), pp. 340–353. Available at: https://doi.org/10.1093/sysbio/syx074.
Budd, G.E. and Mann, R.P. (2022) Two notorious nodes: a critical examination of MCMCTree relaxed molecular clock estimates of the bilaterian animals and placental mammals. preprint. Paleontology. Available at: https://doi.org/10.1101/2022.07.01.498494.
Cunningham, J.A. et al. (2017) ‘The origin of animals: Can molecular clocks and the fossil record be reconciled?’, BioEssays, 39(1), p. e201600120. Available at: https://doi.org/10.1002/bies.201600120.
Donoghue, P.C.J. and Benton, M.J. (2007) ‘Rocks and clocks: calibrating the Tree of Life using fossils and molecules’, Trends in Ecology & Evolution, 22(8), pp. 424–431. Available at: https://doi.org/10.1016/j.tree.2007.05.005.
Drummond, A.J. and Rambaut, A. (2007) ‘BEAST: Bayesian evolutionary analysis by sampling trees’, BMC Evolutionary Biology, 7(1), p. 214. Available at: https://doi.org/10.1186/1471-2148-7-214.
Eaton, K. et al. (2023) ‘Plagued by a cryptic clock: insight and issues from the global phylogeny of Yersinia pestis’, Communications Biology, 6(1), p. 23. Available at: https://doi.org/10.1038/s42003-022-04394-6.
Fernández, R. et al. (2017) ‘The Opiliones tree of life: shedding light on harvestmen relationships through transcriptomics’, Proceedings of the Royal Society B: Biological Sciences, 284(1849), p. 20162340. Available at: https://doi.org/10.1098/rspb.2016.2340.
Hipsley, C.A. and Müller, J. (2014) ‘Beyond fossil calibrations: realities of molecular clock practices in evolutionary biology’, Frontiers in Genetics, 5. Available at: https://doi.org/10.3389/fgene.2014.00138.
Ho, S.Y.W. et al. (2015) ‘Biogeographic calibrations for the molecular clock’, Biology Letters, 11(9), p. 20150194. Available at: https://doi.org/10.1098/rsbl.2015.0194.
Jiao, X., Flouri, T. and Yang, Z. (2021) ‘Multispecies coalescent and its applications to infer species phylogenies and cross-species gene flow’, National Science Review, 8(12), p. nwab127. Available at: https://doi.org/10.1093/nsr/nwab127.
Kimura, M. (1968) ‘Evolutionary Rate at the Molecular Level’, Nature, 217(5129), pp. 624–626. Available at: https://doi.org/10.1038/217624a0.
Kingman, J.F.C. (1982) ‘The coalescent’, Stochastic Processes and their Applications, 13(3), pp. 235–248. Available at: https://doi.org/10.1016/0304-4149(82)90011-4.
Larget, B. and Simon, D.L. (1999) ‘Markov Chasin Monte Carlo Algorithms for the Bayesian Analysis of Phylogenetic Trees’, Molecular Biology and Evolution, 16(6), pp. 750–759. Available at: https://doi.org/10.1093/oxfordjournals.molbev.a026160.
Lee, M.S.Y. and Palci, A. (2015) ‘Morphological Phylogenetics in the Genomic Age’, Current Biology, 25(19), pp. R922–R929. Available at: https://doi.org/10.1016/j.cub.2015.07.009.
Marris, E. (2004) ‘Molecular clock tied to fossil record’, Nature, pp. news041011-2. Available at: https://doi.org/10.1038/news041011-2.
Martinez, P. (2018) ‘The Comparative Method in Biology and the Essentialist Trap’, Frontiers in Ecology and Evolution, 6, p. 130. Available at: https://doi.org/10.3389/fevo.2018.00130.
Matschiner, M. (2019) ‘Selective Sampling of Species and Fossils Influences Age Estimates Under the Fossilized Birth–Death Model’, Frontiers in Genetics, 10, p. 1064. Available at: https://doi.org/10.3389/fgene.2019.01064.
May, M.R. et al. (2021) ‘Inferring the Total-Evidence Timescale of Marattialean Fern Evolution in the Face of Model Sensitivity’, Systematic Biology. Edited by R. Folk, 70(6), pp. 1232–1255. Available at: https://doi.org/10.1093/sysbio/syab020.
Mott, T. and Vieites, D.R. (2009) ‘Molecular phylogenetics reveals extreme morphological homoplasy in Brazilian worm lizards challenging current taxonomy’, Molecular Phylogenetics and Evolution, 51(2), pp. 190–200. Available at: https://doi.org/10.1016/j.ympev.2009.01.014.
O’Dea, R.E. et al. (2021) ‘Preferred reporting items for systematic reviews and meta‐analyses in ecology and evolutionary biology: a PRISMA extension’, Biological Reviews, 96(5), pp. 1695–1722. Available at: https://doi.org/10.1111/brv.12721.
dos Reis, M., Donoghue, P.C.J. and Yang, Z. (2016) ‘Bayesian molecular clock dating of species divergences in the genomics era’, Nature Reviews Genetics, 17(2), pp. 71–80. Available at: https://doi.org/10.1038/nrg.2015.8.
Ritchie, A.M., Hua, X. and Bromham, L. (2022) ‘Investigating the reliability of molecular estimates of evolutionary time when substitution rates and speciation rates vary’, BMC Ecology and Evolution, 22(1), p. 61. Available at: https://doi.org/10.1186/s12862-022-02015-8.
Ronquist, F. et al. (2012) ‘A Total-Evidence Approach to Dating with Fossils, Applied to the Early Radiation of the Hymenoptera’, Systematic Biology, 61(6), pp. 973–999. Available at: https://doi.org/10.1093/sysbio/sys058.
Rota, J. et al. (2018) ‘A simple method for data partitioning based on relative evolutionary rates’, PeerJ, 6, p. e5498. Available at: https://doi.org/10.7717/peerj.5498.
Wang, Z., Gerstein, M. and Snyder, M. (2009) ‘RNA-Seq: a revolutionary tool for transcriptomics’, Nature Reviews Genetics, 10(1), pp. 57–63. Available at: https://doi.org/10.1038/nrg2484.
Warnock, R.C.M., Yang, Z. and Donoghue, P.C.J. (2017) ‘Testing the molecular clock using mechanistic models of fossil preservation and molecular evolution’, Proceedings of the Royal Society B: Biological Sciences, 284(1857), p. 20170227. Available at: https://doi.org/10.1098/rspb.2017.0227.
Wortel, M.T. et al. (2023) ‘Towards evolutionary predictions: Current promises and challenges’, Evolutionary Applications, 16(1), pp. 3–21. Available at: https://doi.org/10.1111/eva.13513.
Wu, X. and Schepartz, L.A. (2009) ‘Application of computed tomography in paleoanthropological research’, Progress in Natural Science, 19(8), pp. 913–921. Available at: https://doi.org/10.1016/j.pnsc.2008.10.009.
Yates, F. and Healy, M.J.R. (1964) ‘How Should we Reform the Teaching of Statistics?’, Journal of the Royal Statistical Society. Series A (General), 127(2), p. 199. Available at: https://doi.org/10.2307/2344003.
Zuckerkandl, E. and Pauling, L. (1965) ‘Evolutionary Divergence and Convergence in Proteins’, in Evolving Genes and Proteins. Elsevier, pp. 97–166. Available at: https://doi.org/10.1016/B978-1-4832-2734-4.50017-6
Comments